WikiDer > Окисление карбонила реагентами гипервалентного йода
Окисление карбонила реагентами гипервалентного йода включает функционализацию α-положения карбонильных соединений через посредство енолятов гипервалентного йода (III). Этот электрофильный промежуточный продукт может подвергаться атаке различных нуклеофилов или подвергаться перегруппировке или элиминированию.[1]
Вступление
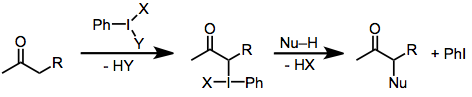
Соединения гипервалентного йода (III) являются привлекательными окислителями из-за их стабильности и селективности. В присутствии енолизируемых карбонильных соединений они способны осуществлять окислительную функционализацию α-положения. Ключевые промежуточные формы енолята йода (III), которые затем подвергаются нуклеофильному замещению (α-функционализация), элиминированию (дегидрированию) или перегруппировке. Общие реагенты гипервалентного йода, используемые для осуществления этих превращений, включают: йодозилбензол (PhIO),[2] иодобензолдиацетат (PhI (OAc)2),[3] Реактив Козера (PhI (OTs) OH),[4] и (дихлоридо) бензол (PhICl2).[5]
(1)

Механизм и стереохимия
Преобладающий механизм
Механизм окисления карбонила реагентами йода (III) варьируется в зависимости от структуры субстрата и условий реакции, но возможны некоторые обобщения. В основных условиях активные йодирующие соединения представляют собой соединения йода (III), в которых любые относительно кислые лиганды на йоде (такие как ацетат) заменены алкоксидом.[2] Во всех случаях α-углерод образует связь с йодом. Восстановление йода (III) до йода (I) затем происходит через атаку нуклеофила на теперь электрофильный α-углерод. В основных условиях нуклеофильная атака карбонильного углерода происходит быстрее, чем атака альфа-углерода. Замещение йода фактически осуществляется внутримолекулярно карбонильным кислородом, который становится α-гидроксильным кислородом в продукте.[6]

(2)

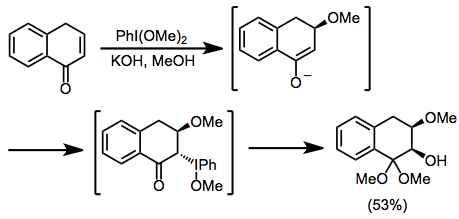
Наблюдались перегруппировки енолятов йода (III). В кислых условиях окисление ариленольных эфиров приводит к α-ариловым эфирам за счет миграции 1,2-арила.[7] Сжимающие кольцо перегруппировки Фаворского могут иметь место при основных условиях (см. Уравнение (12) ниже).
(3)

Стереохимия
Используя карбонильный комплекс хрома, было показано, что замещение йода, вероятно, происходит с инверсией конфигурации. Йод приближается к стороне, противоположной трикарбонильному звену хрома, из-за стерических затруднений. Обратное смещение приводит к син связь между хромом и α-гидроксильной группой.[8]

(4)

Исследования окисления ненасыщенных карбонильных соединений также дают представление о стереохимии. Только изомер с син наблюдалась взаимосвязь между α-гидрокси и β-метоксигруппами. После нуклеофильной атаки метоксидом йод приближается к поверхности, противоположной метоксиду. Обратное вытеснение гидроксидом затем приводит к син изомер.[9]
(5)

Объем и ограничения
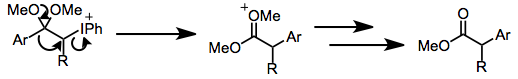
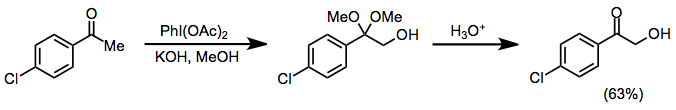
В протонных условиях кетоны подвергаются α-гидроксилированию и образованию диметилацеталя. Иодозилбензол, и иодбензолдиацетат (IBD) могут влиять на это превращение. Этот метод может быть использован для синтеза α-гидроксикетонов после кислотного гидролиза кетальной функциональности.[10]
(6)

В присутствии солей диарилиодония еноляты подвергаются α-арилированию. Объемные диарилиодониумы реагируют медленнее и енолируют гомосцепление (см. Уравнение (10) ниже) начинает конкурировать при замещении ароматического кольца.[11]

(7)

α-Окситозилирование способствует превращению карбонильных соединений в различные α-функционализированные продукты. Образующиеся α-тозилоксикарбонильные соединения более стабильны, чем α-галогенкарбонильные соединения, и не являются лакриматорами.[12]

(8)

Простые эфиры силиленола претерпевают многие из тех же реакций, что и карбонильные соединения, в присутствии реагентов йода (III). α-Алкоксилирование возможно в присутствии внешнего нуклеофила спирта, хотя выходы несколько изменяются.[13]
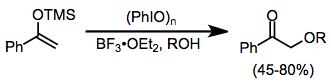
(9)

Когда нет внешнего или внутреннего нуклеофила, происходит окислительное гомосочетание с образованием 1,4-дикарбонильных соединений.[14]

(10)

Внутримолекулярно связанные нуклеофилы могут замещать йодбензол с образованием лактонов или других гетероциклов.[15] Если в циклическом продукте присутствуют кислые водороды, в условиях реакции может происходить переокисление.

(11)

В некоторых случаях перегруппировки затрудняют окисление карбонильных соединений гипервалентным йодом. Арилмиграция может происходить в кислых условиях, давая α-ариловые эфиры из енольных эфиров.[7] Также наблюдались перегруппировки Фаворского, которые были особенно полезны для синтеза стероидов.[16]

(12)

Синтетические приложения
Окислительная функционализация простых эфиров силиленола в низкой концентрации (во избежание гомосцепления) без внешнего нуклеофила приводит к дегидрированию. Это может быть полезным способом получения α, β-ненасыщенных карбонильных соединений при отсутствии функциональных маркеров. Например, дегидрирование используется в синтезе стероидов для образования ненасыщенных кетонов.[17]
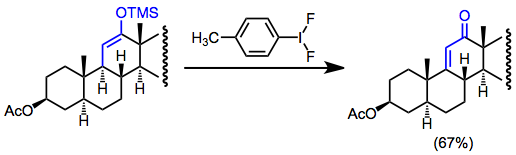
(13)

Сравнение с другими методами
Немногие соединения, окисляющие карбонильные соединения, могут соперничать по безопасности, селективности и универсальности с реагентами на основе гипервалентного йода. В других методах α-гидроксилирования карбонильных соединений могут использоваться токсичные металлоорганические соединения (такие как тетраацетат свинца или четырехокись осмия). Одной альтернативой гипервалентному окислению йода, в котором не используются тяжелые металлы, является воздействие енолята металла на дикислород с последующим восстановлением образующегося пероксида (уравнение (14)). Наиболее популярным методом α-гидроксилирования карбонильных соединений является Руботтовое окисление, который использует простые эфиры силил енола в качестве субстратов и перкислоты в качестве окислителей.[18]
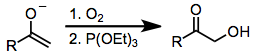
(14)

Окислительные перегруппировки обычно легче осуществить с использованием реагентов на основе гипервалентного йода, чем с использованием других окислителей. Например, реакция Вилльгеродта-Киндлера алкиларилкетонов требует принудительных условий и часто дает низкие выходы амидных продуктов.

(15)

Условия и порядок экспериментов
Пример процедуры[19]

(16)

Гидрокси (мезилокси) иодбензол (3,16 г, 10 ммоль) добавляли к раствору метилтриметилсилилфенилкетенацеталя, полученного из метилфенилацетата (3,33 г, 15 ммоль), в сухом дихлорметане (50 мл). Смесь перемешивали при комнатной температуре в течение 2 часов, а затем промывали водным раствором бикарбоната натрия (3 × 50 мл). Органическую фазу сушили (MgSO4) и концентрировали в вакууме с получением сырого мезилоксиэфира, который очищали колоночной хроматографией на силикагеле (гексан-дихлорметан, 1: 1), получая 1,58 г (65%) указанного в заголовке соединения, т.пл. 91-92 °; ИК (KBr) 1760 см−1 (CO); 1H ЯМР (CDCl3): δ 3,10 (с, 3H), 3,80 (с, 3H), 6,00 (с, H), 7,40-7,80 (м, 5H); 13C ЯМР (CDCl3): δ 168,2 (с), 132,2 (с), 130,0 (с), 129,0 (с), 127,7 (с), 78,9 (с), 53,0 (с), 39,45 (с); МС, m / z 185 (53), 165 (15), 145 (15), 107 (100), 90 (12), 79 (65), 51 (17).
Рекомендации
- ^ Мориарти, Р. М .; Пракаш, О. Орг. Реагировать. 1999, 54, 273. Дои:10.1002 / 0471264180.or054.02
- ^ а б Schardt, B.C .; Хилл, К. Л. Неорг. Chem. 1983, 22, 1563.
- ^ Мориарти, Р. М .; Хм. Tetrahedron Lett. 1981, 22, 2747.
- ^ Koser, G.F .; Реленый, А.Г .; Kalos, A. N .; Ребрович, Л .; Веттах, Р. Х. J. Org. Chem. 1982, 47, 2487.
- ^ Днепровский, А. С .; Крайнюченко, И. В .; Темникова, Т.И. J. Org. Chem. СССР (англ. Пер.) 1978, 14, 1414.
- ^ Мориарти, Р. М .; Хм.; Гупта, С. Tetrahedron Lett. 1981, 22, 1283.
- ^ а б Пракаш, О. Aldrichimica Acta 1995, 28, 63.
- ^ Мориарти, Р. М .; Engerer, S.C .; Пракаш, О .; Пракаш, I .; Gill., U. S .; Фриман, В.А. J. Org. Chem. 1987, 52, 153.
- ^ Tamura, Y .; Якура, Т .; Terashi, H .; Haruta, J .; Кита, Ю. Chem. Pharm. Бык. 1987, 35, 570.
- ^ Подолесов, Б. J. Org. Chem. 1984, 49, 2644.
- ^ Берингер, Ф. М .; Гальтон, С.А. J. Org. Chem. 1963, 28, 3417.
- ^ Пракаш, О .; Гоял, С. Синтез 1992, 6291.
- ^ Мориарти, Р. М .; Пракаш, О .; Duncan, M. P .; Vaid, R.K .; Мусаллам, Х.А. J. Org. Chem. 1987, 52, 150.
- ^ Мориарти, Р. М .; Пракаш, О .; Дункан, М. П. J. Chem. Soc., Chem. Commun. 1985, 420.
- ^ Мориарти, Р. М .; Пракаш, О .; Пракаш, I .; Мусаллам, Х.А. J. Chem. Soc., Chem. Commun. 1984, 1342.
- ^ Даум, С. Дж. Tetrahedron Lett. 1984, 25, 4725.
- ^ Цусима, Т .; Kawada, K .; Цудзи, Т. Tetrahedron Lett. 1982, 23, 1165.
- ^ Chen, B.-C .; Чжоу, П .; Дэвис, Ф. А .; Цыганек, Э. Орг. Реагировать. 2003, 62, 1.
- ^ Мориарти, Р. М .; Penmasta, R .; Awasthi, A.K .; Epa, R.W .; Пракаш, И. J. Org. Chem. 1989, 54, 1101.